by
and

Biophysica 2023, 3(3), 476-484; https://doi.org/10.3390/biophysica3030031 - 26 Jul 2023
Abstract
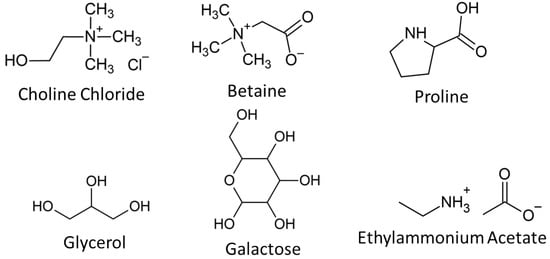
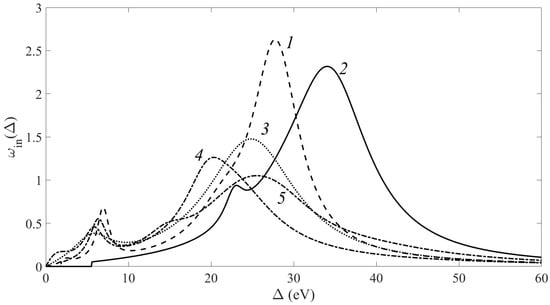
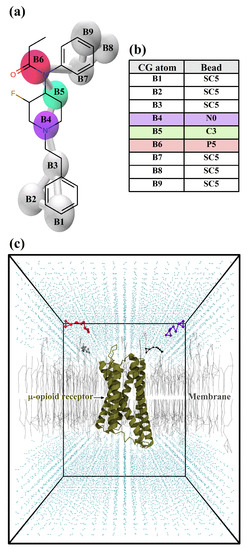
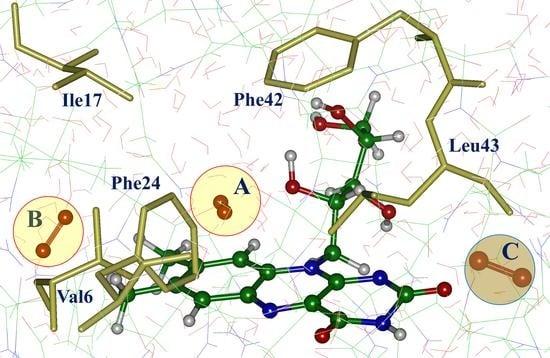
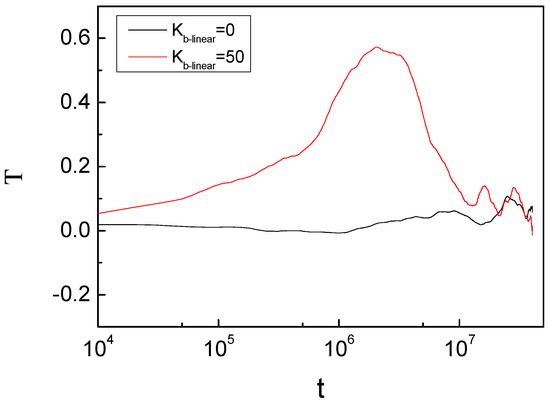
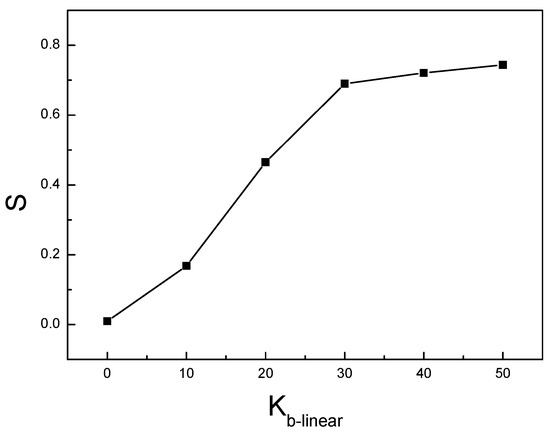
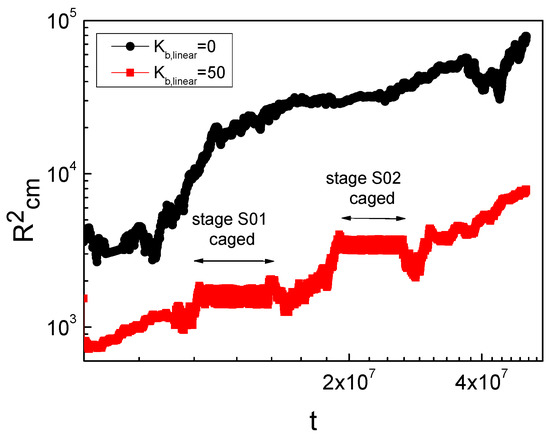
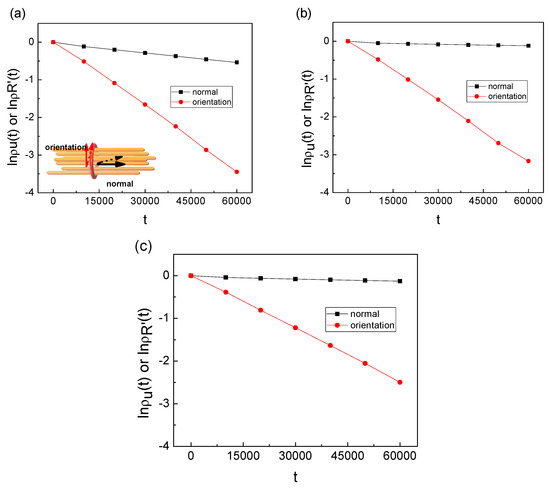
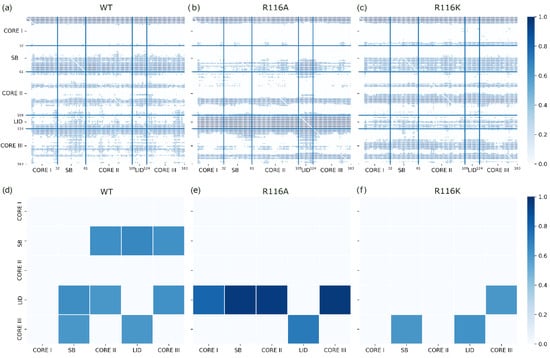
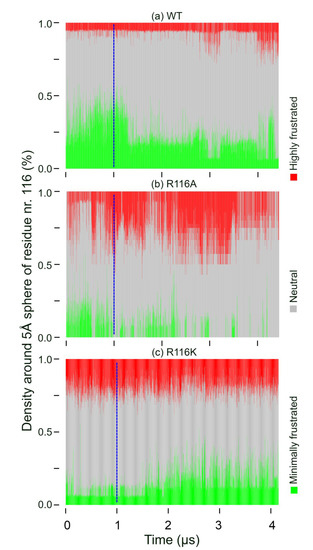
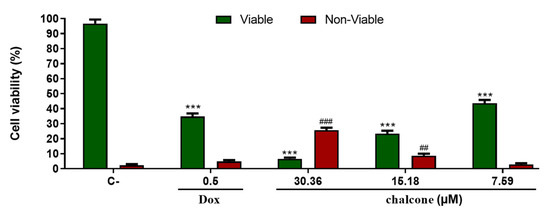
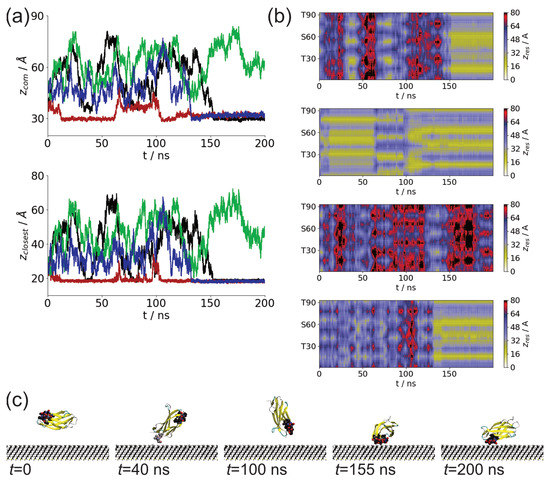
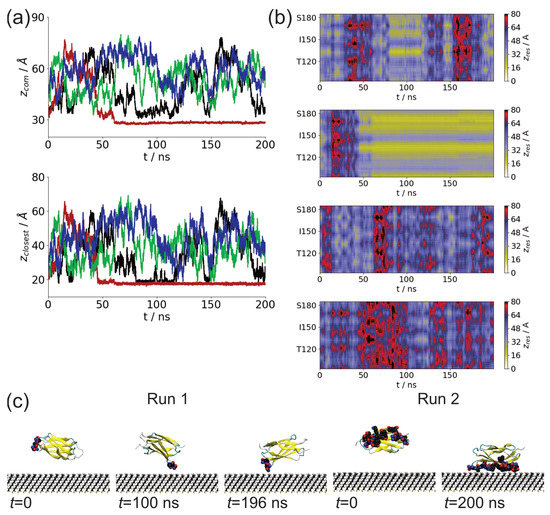
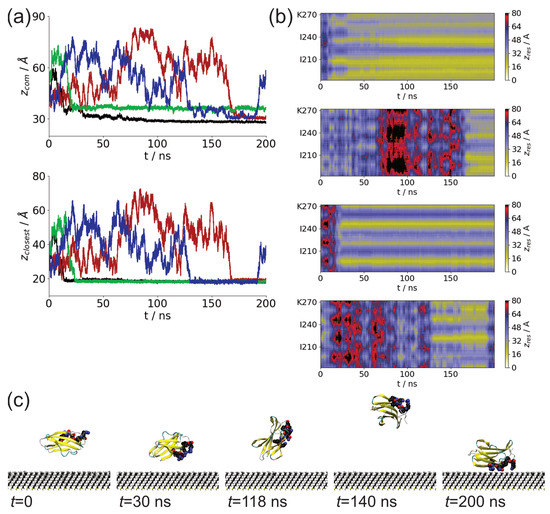
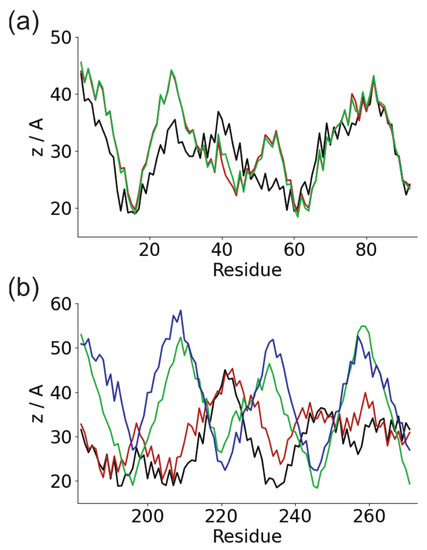
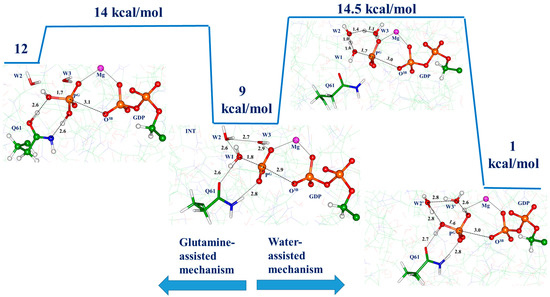
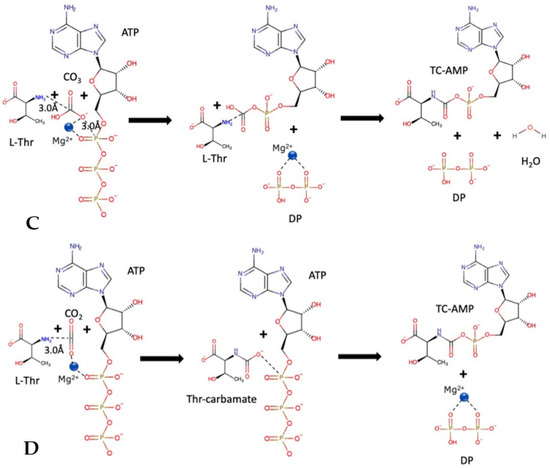
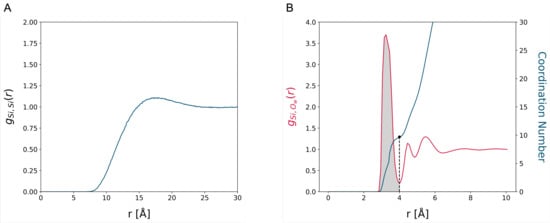
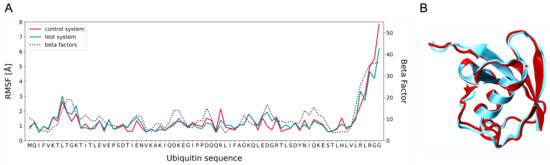
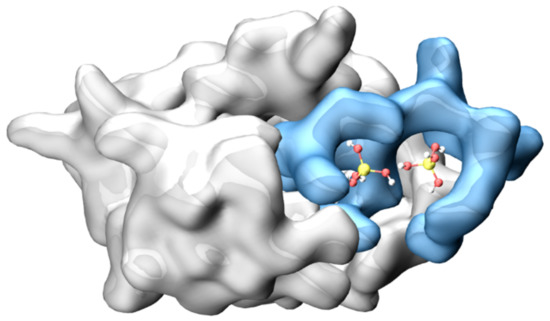
We studied the dynamic behavior of a single semiflexible ring in linear chain matrix based on a coarse-grained model using the molecular dynamics simulation approach. We found that that ring chains’ hollow centers are frequently filled with linear chains. However, as the rigidity
[...] Read more.
We studied the dynamic behavior of a single semiflexible ring in linear chain matrix based on a coarse-grained model using the molecular dynamics simulation approach. We found that that ring chains’ hollow centers are frequently filled with linear chains. However, as the rigidity of the linear chains increases, the linear chains arranged parallel to each other and the ring chain are temporary caged. As a result, the swing movement in the normal direction of the ring is significantly limited, and the relaxation time in the normal direction increases significantly. Our findings can help to understand the physical mechanism of the movement of the ring chain in ring–linear polymer blends at the microscopic level.
Full article
(This article belongs to the Special Issue Molecular Structure and Simulation in Biological System)
►
Show Figures
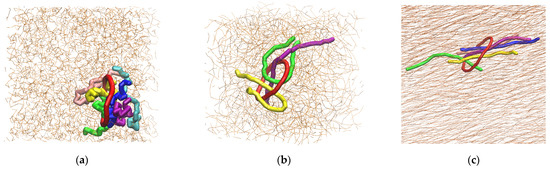
Figure 1











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}